We have seen examples of how patients with ‘normal’ thyroid profiles (TSH, fT3, fT4 each within its reference interval) are severely hypothyroid. From casual glances at thyroid support group fora I have seen over 50 such examples. When diagnosing and treating hypothyroidism there is a need for good science not dogmatic practice founded on over-simplified models of thyroid function.
TSH, fT3, fT4 and TRH are Interdependent
TSH, fT3, fT4 and TRH work as a system, they are interdependent and cannot be interpreted as if they are independent. It’s not sufficient that each hormone falls within its reference interval, the normal relationship (the axis) must function correctly. If TSH, fT3 and fT4 are all low normal then something is wrong – it isn’t the way the axis works.
TSH can be a valuable tool if interpreted with care. For example, primary hypothyroidism can be detected at an early stage. Normal TSH (or fT3 or fT4) does not exclude hypothyroidism. Before relying on thyroid function tests (TFTs) we must ensure the thyroid axis is working correctly. When the axis isn’t functioning correctly TFT interpretation becomes complex.
TSH bioactivity is influenced by illness and TRH stimulation. TSH immuno-assays measure the presence of molecules, they don’t measure bioactivity. ‘a graveyard has a high density of people with little activity‘. A subnormal TSH due to insufficient TRH stimulation has low bioactivity.
Serum hormone does not always reflect intracellular levels. To get an insight into thyroid status we have to ask ‘where is the T3 coming from?’.
Thyroid status is determined by T3 saturation at receptors. In organs such as the brain, T3 comes primarily from type-2 deiodinase (D2) converting T4 to T3 locally, serum T3 has a minor role.
D2 is the major source of serum T3 in euthyroid individuals and even more so in hypothyroidism. A reduction in D2 activity has a devastating effect on intracellular T3. It is sometimes asserted ‘T4 isn’t entering the cells‘. T4 is entering the cells but if insufficient T3 is generated locally (by D2) there will be less T3 leaving the cells resulting in lower fT3 levels. T3 comes from cells, from thyroid cells or deiodinase activity within cells (or from tablets).
TSH stimulates D2. If TSH is subnormal (fT3, fT4 not elevated) intracellular T3 will be low and the patient hypothyroid. The patient is hypothyroid because their TSH is not elevated, it has low bioactivity and fails to stimulate sufficient secretion and deiodinase.
The importance of D2 in the brain
D2 regulates T3 in several organs including the brain. The brain controls patients’ perception of thyroid status, cognitive functioning and influences other organs. Doctors see blood hormone levels, patients experience brain hypothyroidism, this results in conflict between doctors and patients.
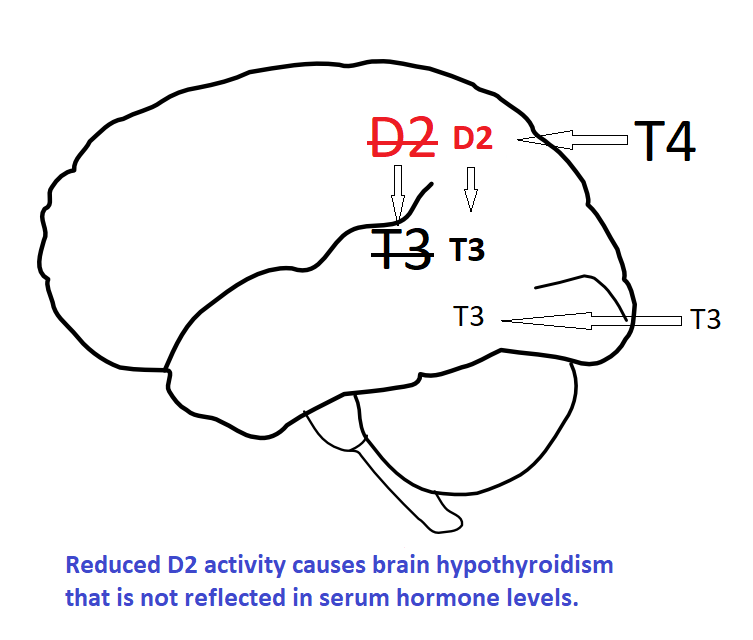
There is temptation to assume that if we restore serum T3 levels to normal then normal tissue levels will follow. This is not so, tissues use T4 and D2 to regulate local T3. If D2 activity is impaired normal serum hormone levels will not result in normal tissue levels. Supra-physiological serum T3 is needed to override normal regulation and provide sufficient local T3. This may result in too much T3 for tissues not subject to local regulation by D2 and D3. Serum is the intermediate space; thyroid status is determined by local T3 not circulating T3.
Consider an analogy. In cases of dehydration or renal disease a nurse may measure 24-hour urine volume to assess kidney function. If the volume is low the nurse could ‘correct’ the result by adding some urine to the container. The numbers are corrected but the patient’s status is not. The same logic applies to ‘correcting’ serum T3, the numbers look good but the local T3 status will still be deficient. What matters is ‘where the T3 is coming from‘.
How can we determine thyroid status in the brain?
Measuring thyroid hormone status in the brain directly is difficult, inserting probes or taking biopsies would not be popular! We could carry out cognitive testing, this has been done and shows impairment in some patients e.g. those with D2 polymorphisms. This has limited success, perhaps because it is not targeted, patients have not been asked what problems they experience. Personally, I find most activities are OK but if I try to concentrate for a sustained period (e.g. reading studies) I feel tired and can no longer focus. It’s not a question of being able to concentrate but able to sustain concentration over a period of time. (Perhaps T4 in the brain acts as a reservoir for such tasks?) Current cognitive testing has not identified or targeted appropriate factors.
A research suggestion
I notice that if I get my L-T3 dose right my sleep is deeper with more vivid dreams and this leads to improved cognitive function the following day. Hypothyroidism is associated with impaired sleep quality with less deep stage 3 and stage 4 sleep, this is corrected by desiccated thyroid as seen in the following study.

A prospective randomised placebo-controlled study of L-T3 treatment of symptomatic patients who meet the criteria of subnormal TSH should be conducted. Sleep monitoring could be used as a model for assessment of such patients, to determine whether they have abnormal sleep patterns and if they respond to L-T3 therapy. This would also determine the L-T3 dose required to restore normal thyroid status in the brain. A follow-up study could look at the effect of exogenous TRH in a similar manner.
The balance of evidence
A large body of thyroid research is substandard, based on the false assumption that hypothyroidism requires a raised TSH and low fT4 (except rare cases of central hypothyroidism). Good research is driven by observation (e.g. patient experience) not dogmatic reliance on TFTs. It’s an appropriate time to remember the principles of good science as expressed by our old friend Richard Feynman. Large numbers of patients have severe hypothyroidism with ‘normal’ TFTs which responds to thyroid hormone therapy with a high proportion of T3. Evidence is clinical response, not ‘expert’ opinion.
This topic contains a number of assertions so we will look at the balance of evidence for each.
Assertion: Subnormal TSH exists and is not just a normal day to day hormonal variation. There’s good evidence for this. We know it happens in non-thyroidal illness (euthyroid sick syndrome) and low-T3 syndrome. The hypothalamus pituitary axis can be down-regulated.
Actual examples of a down-regulated axis in patients are rare because TFTs are not carried out before patients become hypothyroid. However, my hypothyroidism was unusual, I have my TFTs from before therapy and again after a decade of TSH suppression demonstrating a down-regulated axis.
There is a large body of anecdotal evidence from patients with the characteristic subnormal TSH profile who present with hypothyroid signs and symptoms which respond to thyroid hormone therapy. A prospective placebo-controlled trial would give formal confirmation. A study of TSH bioactivity in this group of patients might find subnormal TSH secretion.
Experimental and population studies show that lowering fT3 or fT4 raises TSH, in this situation a non-elevated TSH is abnormal.
Assertion: D2 is crucial to thyroid status. Seventy percent of serum T3 is derived from D2 conversion of T4 to T3. D2 and D3 regulate intracellular T3 and hence thyroid hormone action. The brain derives most of its T3 from D2 activity and is relatively insensitive to changes in serum T3.
Assertion: TSH promotes deiodinase. Many experiments have demonstrated that deiodinase is regulated by TSH as well as by thyroid hormones and other factors. Tissues express TSH receptors and deiodinase in these tissues is subject to TSH stimulation. In hypothyroidism deiodinase increases if TSH is elevated but not if TSH is low. In Graves’ disease TSH receptor antibodies stimulate deiodinase activity.
TSH has isoforms with varying potency, TSH bioactivity increases as TRH stimulation increases. TSH has thyroid stimulating and deiodinase stimulating properties.
On the other hand, a study with recombinant human TSH found an inverse relationship between rhTSH and thyroid hormone. rhTSH is not TSH and may not have deiodinase promoting properties. A further complication is serum hormone levels were measured three days after rhTSH was injected. A study using TRH to promote TSH might clarify this issue.
Some hypothyroid patients report that L-T4 paradoxically makes them worse. In subnormal TSH L-T4 will suppress TSH and further reduce deiodinase activity resulting in lower intracellular T3.
Assertion: Low intracellular T3 caused by subnormal TSH will be reflected in low serum T3. This makes sense but seeking evidence is tricky. We have some understanding of how hormone is transported into cells and have identified some of the transport proteins. I’m not aware of studies that investigate how T3 and T4 are transported out of cells, or how this outflow is regulated. It’s theoretically possible that the cell would hold onto T3 if it were deficient and so serum T3 would exaggerate cellular deficiency. It’s also possible there is no such regulation, that the exit rate of T3 from the cell is constant. To the best of my knowledge this is an unknown. On the other hand, we could argue that the evidence from patients is that intracellular T3 is low as they have pronounced signs and symptoms of low intracellular T3 such as severe tiredness, cognitive impairment and muscular pain or fibromyalgia. More conclusive evidence would be welcome.
We know that T3 comes from D2 activity and tissues release D2 generated T3 in varying amounts. For example, when subjects are exposed to cold D2 activity in brown fat (BAT) drastically increases. However, if we measure serum fT3 before and after cold exposure we see a small increase. Clearly in this circumstance D2 derived T3 in BAT makes a minor contribution to serum T3. It seems that low serum fT3 levels reflect low intracellular levels but we do not know to what extent and in which tissues.
Time for a fundamental change in approach
There is clear evidence that hypothyroidism with normal TFTs exists and is not rare. The pretence that it does not exist must stop.
We have seen that hypothyroidism with normal blood hormone levels exists and that restoring normal serum hormone levels may not be sufficient. There are consequences of a down-regulated axis so it makes sense to avoid suppressing TSH if possible.
Help from patients
If you find this topic helpful and feel it makes sense, please consider sharing it with others. We need to raise awareness of the various causes of hypothyroidism, especially those that present with blood hormone levels that appear ‘normal’.